
PART ONE OF A TWO-PART SERIES
By Margaret Bobonich, DNP, FNP-C, DCNP, FAANP
Abstract
Bullous pemphigoid (BP) is the most common autoimmune blistering disease in the elderly, marked by subepidermal blistering and considerable morbidity. Diagnostic delays are common due to its clinical variability and immunopathologic complexity, particularly in early or atypical cases.
This review highlights key aspects of BP, including its pathogenesis, clinical variants, and diagnostic strategies. Emphasis is placed on distinguishing features of BP and the importance of integrating histopathology, direct and indirect immunofluorescence, and serologic testing to improve diagnostic accuracy. As BP incidence rises with aging populations and polypharmacy, increased clinical awareness is essential to optimize outcomes.
- Introduction
- Pathogenesis
- Prognosis and Comorbidities
- Clinical Presentation
- Differential Diagnosis
- Diagnostic Workup
- Conclusion
Introduction
Bullous pemphigoid (BP) is the most prevalent autoimmune subepidermal blistering disease exhibiting a strong predilection for elderly individuals. The annual incidence of BP ranges from 2.4 to 23 cases per million people annually, with rates exceeding 300 per million in those aged 80 years and older.1,2 The increasing incidence has been attributed to an increasing life expectancy, improved recognition of atypical clinical features, enhanced availability of diagnostic modalities (serologic and immunofluorescence assays), and the growing burden of polypharmacy among older adults.3,4 Early recognition and accurate diagnosis are essential to mitigate disease progression and reduce associated morbidity.
Although most cases of BP are idiopathic, a range of comorbid conditions and medications have been implicated in the onset of both bullous and non-bullous variants. Among the drug classes, the strongest association has been reported with dipeptidyl peptidase-4 inhibitors (gliptins), particularly in elderly patients with Type 2 diabetes mellitus.5 Additional drugs implicated include immune checkpoint inhibitors (e.g., PD-1/PD-L1 inhibitors), loop diuretics, non-steroidal anti-inflammatory drugs (NSAIDs), penicillins, antimicrobials, thiazides, biologics, and certain vaccines.6 The pathogenesis of drug-associated bullous pemphigoid (DABP) is thought to involve multiple mechanisms, including unmasking of genetic susceptibility, immune dysregulation, and structural alteration of antigenic targets.5,7 Recognizing drugs commonly associated with BP is essential for clinicians to identify cases of DABP promptly and to consider discontinuation of the suspected agent.
Pathogenesis
The pathogenesis of BP is primarily mediated by an autoimmune response against structural components of the dermoepidermal junction—specifically, the hemidesmosomal proteins BP180 (type XVII collagen) and BP230 (Figure 1A). These proteins are critical for maintaining adhesion between the basal keratinocytes and the underlying basement membrane zone (BMZ).8-10 Autoantibodies—primarily of the IgG class—target these proteins, initiating a cascade of inflammatory events that ultimately result in subepidermal blister formation (Figure 1B). In addition to IgG autoantibodies, IgE antibodies against BP180 have been identified in a subset of patients and are associated with mast cell degranulation, histamine release, and enhanced inflammatory responses.11
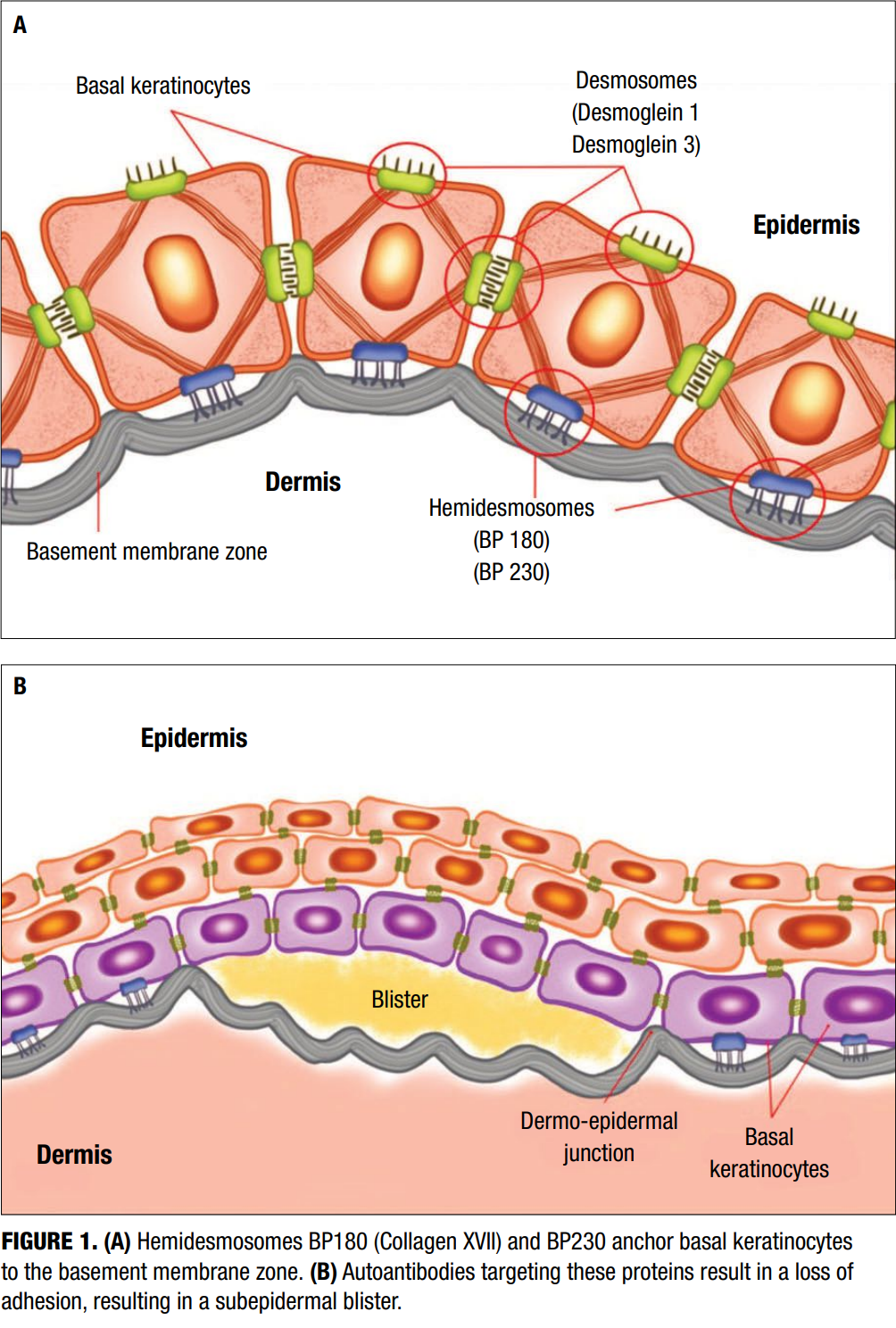
Upon binding of autoantibodies to BP180 and BP230, the classical complement pathway is activated, leading to the deposition of C3, which plays a central role in modulating inflammation. These components promote the recruitment of neutrophils, eosinophils, and mast cells, which release proteolytic enzymes (e.g., matrix metalloproteinases) that degrade anchoring filaments and other BMZ components, thereby inducing separation of the dermoepidermal junction (Figure 1).12,13 A strong Th2 inflammatory immune response characterizes the immunologic milieu in BP. Increased levels of interleukin (IL)-4 and IL-13 enhance the production of IgE and support eosinophil recruitment and survival, thereby amplifying the inflammatory response.14 Additionally, IL-5 plays a key role in eosinophil activation, and IL-17 may contribute to early neutrophilic infiltration. Recent studies have also implicated regulatory T-cell dysfunction and epitope spreading as contributing factors to chronicity and disease progression.15 The interplay between autoantibody production, cellular immunity, and the skin’s structural components underscores the complex and multifactorial nature of BP pathogenesis.
Prognosis and Comorbidities
BP is associated with considerable morbidity and a significant risk of mortality, particularly among elderly populations. The 1-year mortality rate has been estimated at approximately 23.5%, a figure influenced by multiple factors including advanced age, disease severity, treatment-related adverse effects, and the presence of comorbid conditions.1 Chronic pruritus, widespread erosions, and secondary infections contribute to substantial physical discomfort and psychological distress, significantly impairing patients’ quality of life.4 In cases involving mucosal membranes—including the oral cavity, nasopharynx, esophagus, genitalia, and rectum—patients may suer from severe pain, scarring, dysphagia, and nutritional compromise due to impaired oral intake.16
“Chronic pruritus, widespread erosions, and secondary infections contribute to substantial physical discomfort and psychological distress, significantly impairing patients’ quality of life.”
Numerous comorbidities have been linked to BP, with a particularly strong association seen with neurologic diseases such as Alzheimer’s disease, Parkinson’s disease, stroke, and multiple sclerosis.2,17 Additionally, patients with BP exhibit a higher prevalence of cardiovascular disease, diabetes mellitus, and renal insuffciency, conditions that may exacerbate disease management challenges and increase vulnerability to complications.18 Of notable concern is the markedly elevated risk—up to 15-fold higher—of venous thromboembolism (VTE), including deep vein thrombosis and pulmonary embolism, particularly during active disease flares or prolonged immobility.19 Given these risks, clinicians should adopt a comprehensive approach to BP management, with vigilant monitoring for comorbidities and proactive efforts to minimize complications. Interdisciplinary care involving dermatology, neurology, cardiology, and geriatrics may enhance outcomes in this vulnerable patient population.
“Numerous comorbidities have been linked to BP, with a particularly strong association seen with neurologic diseases such as Alzheimer’s disease, Parkinson’s disease, stroke, and multiple sclerosis.”
Clinical Presentation
Classic bullous pemphigoid
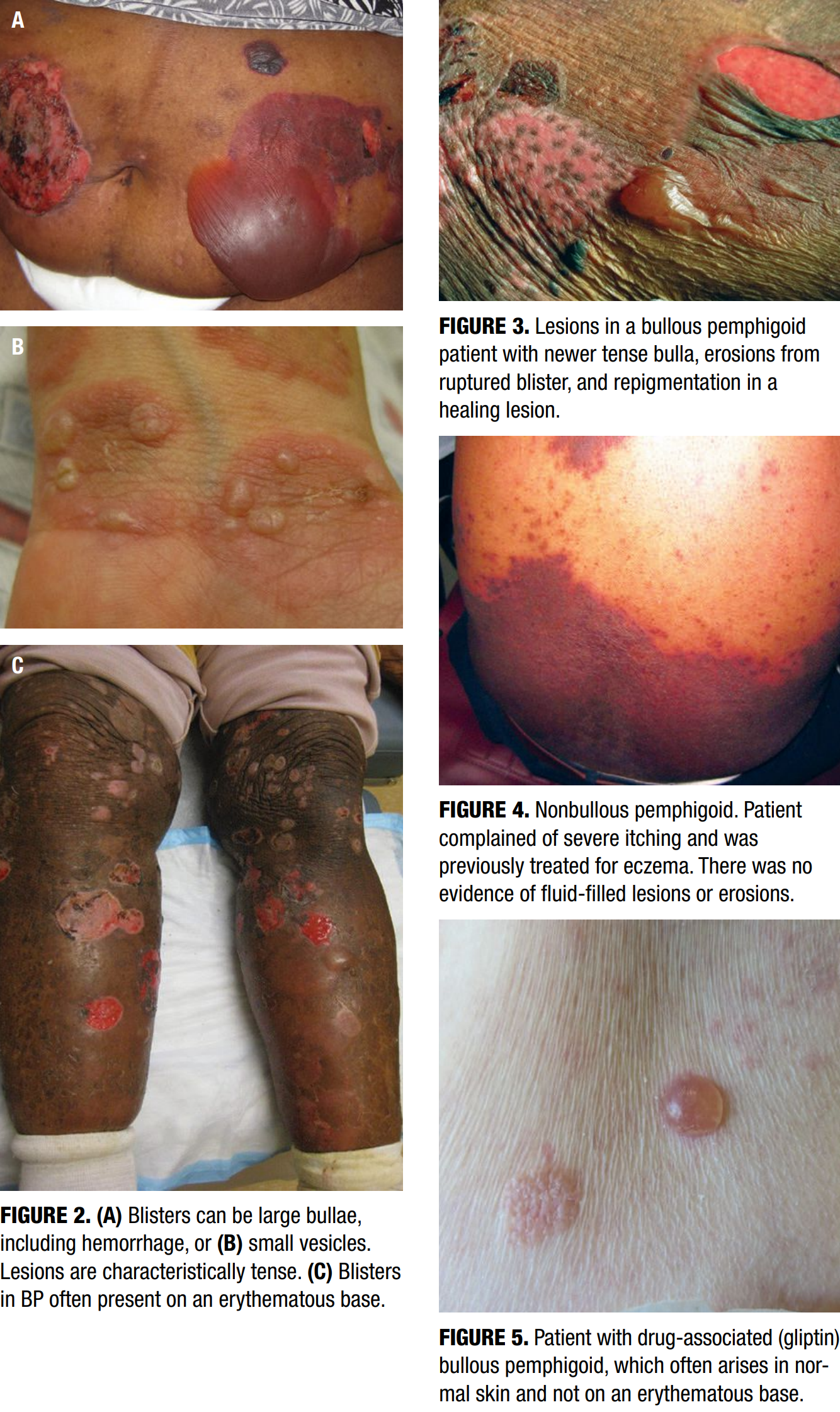
BP classically presents with tense, fluid-filled vesicles and bullae on an erythematous or urticarial base (Figure 2). Pruritus is often severe and may precede the appearance of bullae by several weeks. Blisters are typically symmetrically distributed, with a predilection for the trunk, groin, extremities, and flexural surfaces. Bullae may appear in various stages, including new blisters, erosions, and healing lesions (Figure 3). BP often results in post-inflammatory hyperpigmentation, although scarring is uncommon.8–10
Unlike pemphigus vulgaris, BP is characterized by a negative Nikolsky sign, which reflects the subepidermal level of blister formation. The Nikolsky sign is a useful clinical test to differentiate intraepidermal from subepidermal blistering disorders. A positive Nikolsky sign is demonstrated when gentle lateral pressure is applied to normal-appearing perilesional skin, resulting in shearing away of the epidermis. This sign is typically observed in pemphigus vulgaris, Staphylococcal scalded skin syndrome, and Stevens–Johnson syndrome. In contrast, BP lesions usually resist such shearing due to their deeper dermoepidermal separation.
Mucosal involvement occurs in approximately 10–30% of BP patients and most commonly affects the oral and genital mucosa. Oral lesions may be associated with increased disease severity and are more often seen in patients lacking anti-BP230 autoantibodies.16 The presence of mucosal involvement may result in pain, nutritional compromise, and functional impairment.
Non-bullous phase or non-bullous pemphigoid
Up to 20% of BP patients may initially present in a non-bullous phase, characterized by intensely pruritic urticarial plaques (Figure 4), eczematous patches, papules, or nodules, without visible blistering.20 This prodromal phase may persist for weeks or months, often resulting in diagnostic delays or misdiagnoses as eczema, urticaria, or drug eruptions. Histopathological findings in early lesions may be non-specific, lacking visible subepidermal clefting. Clinicians should maintain a high index of suspicion in older patients with new-onset, treatment-resistant pruritus with or without primary lesions.
Drug-associated bullous pemphigoid
Drug-associated BP, or DABP, can closely resemble idiopathic forms but may exhibit certain distinguishing features. Younger age of onset, lesions arising on normal skin rather than inflamed bases, and atypical morphologies (e.g., erythema multiforme-like lesions) may suggest a drug-associated etiology (Figure 5).21 DABP may also present with mucosal involvement, scarring, and a positive Nikolsky sign. Clues such as recent drug initiation, polypharmacy, and marked serum eosinophilia with prominent eosinophilic infiltrates on biopsy may further support the diagnosis.
Mucous membrane pemphigoid (MMP)
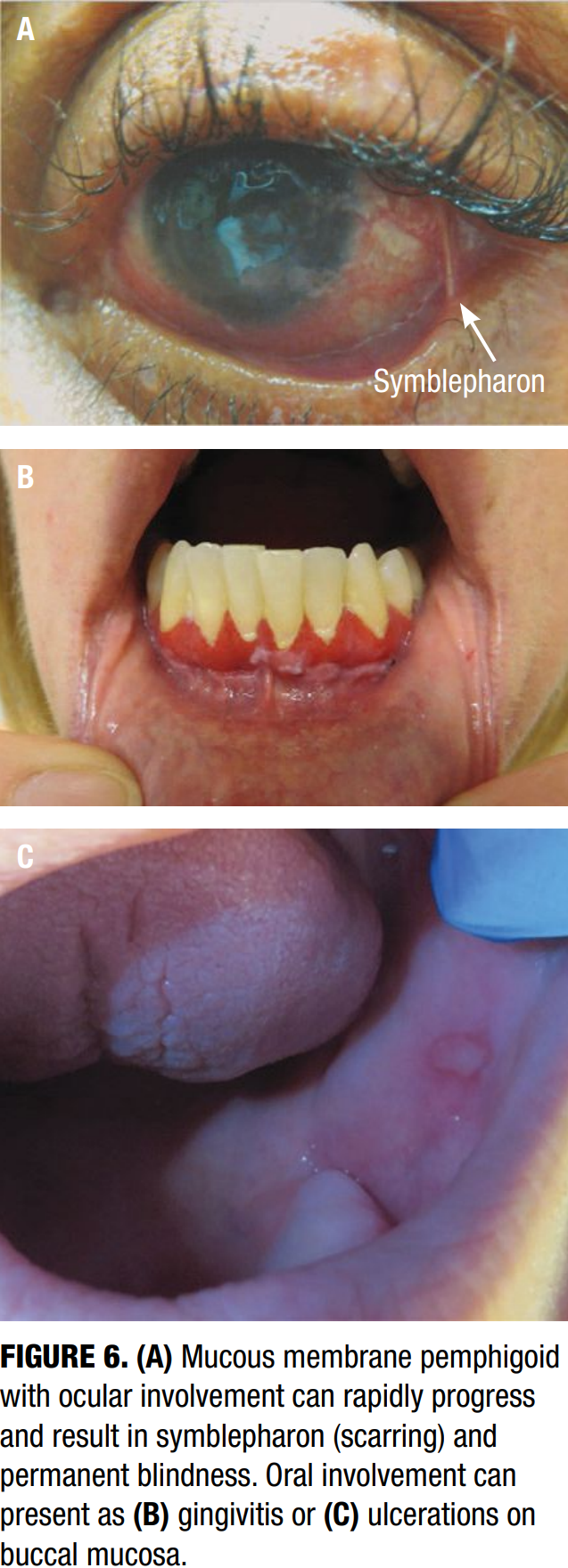
Mucous membrane pemphigoid (MMP) is a rare subtype of pemphigoid that predominantly affects mucosal surfaces—including the oral cavity, conjunctiva, nasopharynx, pharynx, larynx, esophagus, and genital mucosa. Unlike classic BP, MMP frequently causes scarring, which may lead to significant complications such as blindness, airway obstruction, or esophageal strictures.22 Classic MMP presents with erosive gingivitis and genital erosions, while ocular MMP can progress to symblepharon formation and conjunctival fibrosis (Figure 6). Symptoms such as dysphagia or hoarseness may indicate laryngeal or esophageal involvement. Diagnosis relies on direct immunofluorescence, which reveals linear IgG and C3 deposition at the basement membrane zone. Multidisciplinary care involving dermatology, ophthalmology, and otolaryngology is often necessary.
Pemphigoid gestationis (PG)
Pemphigoid gestationis (PG), or herpes gestationis, is a rare autoimmune blistering disorder occurring during pregnancy, most often in the second or third trimester. It typically begins with pruritic urticarial papules and plaques, often starting in the periumbilical region and progressing to vesicles or tense bullae with centrifugal spread.23 Postpartum flares are common and may be triggered by menstruation or oral contraceptives. PG is associated with increased risk for Grave’s disease and other autoimmune conditions. Fetal risks include growth restriction and preterm delivery.24
Differentiating PG from pruritic eruption of pregnancy (PEP) can be challenging, especially during non-bullous phases.25 PEP more commonly occurs in first pregnancies, multiple gestations, and during the late third trimester. Unlike PG, PEP typically begins in the striae and spares the umbilical region. Histology alone may not distinguish between PG and PEP; therefore, direct immunofluorescence is essential for diagnosis when PG is suspected.
Differential Diagnosis
The clinical spectrum of BP overlaps with numerous dermatologic conditions, and its differential diagnosis is largely influenced by whether the presentation is bullous or non-bullous. In its early or non-bullous phase, BP may mimic a range of pruritic dermatoses, including atopic dermatitis, urticaria, drug-induced eruptions, and PEP. These conditions are characterized by erythematous, excoriated, or eczematous lesions without visible blistering, often resulting in diagnostic delays, particularly in older adults.
During the bullous phase, BP must be differentiated from other autoimmune blistering diseases (AIBDs). Key conditions in this category include pemphigus vulgaris, which features flaccid intraepidermal bullae and mucosal involvement; linear IgA, known for its annular or grouped vesicles and linear IgA deposition at the basement membrane; dermatitis herpetiformis, associated with gluten sensitivity and granular IgA deposition in dermal papillae; epidermolysis bullosa acquisita, which presents with trauma-induced blisters and dermal-side binding on salt-split skin; and bullous systemic lupus erythematosus, typically occurring in patients with systemic lupus and characterized by widespread vesiculobullous lesions.8,10,26
Other non-autoimmune conditions should also be considered in the differential diagnosis. These include bullous drug eruptions, which often follow recent medication exposure and may display widespread, irregular blistering; infectious etiologies such as bullous impetigo or herpes simplex virus; and bullous erythema multiforme, distinguished by targetoid lesions and mucosal involvement in more severe cases. Accurate diagnosis relies on clinical pattern recognition, detailed medication history, and confirmatory testing via histopathology and direct immunofluorescence.
Diagnostic Workup
While formal diagnostic criteria for BP have not yet been universally established, accurate diagnosis relies on a combination of clinical evaluation, histopathologic findings, direct immunofluorescence (DIF), indirect immunofluorescence (IIF), and serologic studies. A nuanced understanding of biopsy-site selection, appropriate tissue handling, and specimen transport is essential to optimize diagnostic yield, particularly in AIBDs. The integration of these modalities enables a comprehensive and accurate assessment of suspected BP cases (See infographic on page 40). Among these tools, DIF remains the gold standard, providing the highest sensitivity and specificity for confirming BP. However, optimal diagnostic sensitivity is achieved when multiple techniques are employed synergistically, especially in cases with atypical or non-bullous presentation.8,10 Due to the rarity of AIBDs and rapid scientific advances in BP, dermatology clinicians may be challenged to stay current. It is important for dermatology offices to maintain the necessary diagnostic resources.
Histopathology (H&E)
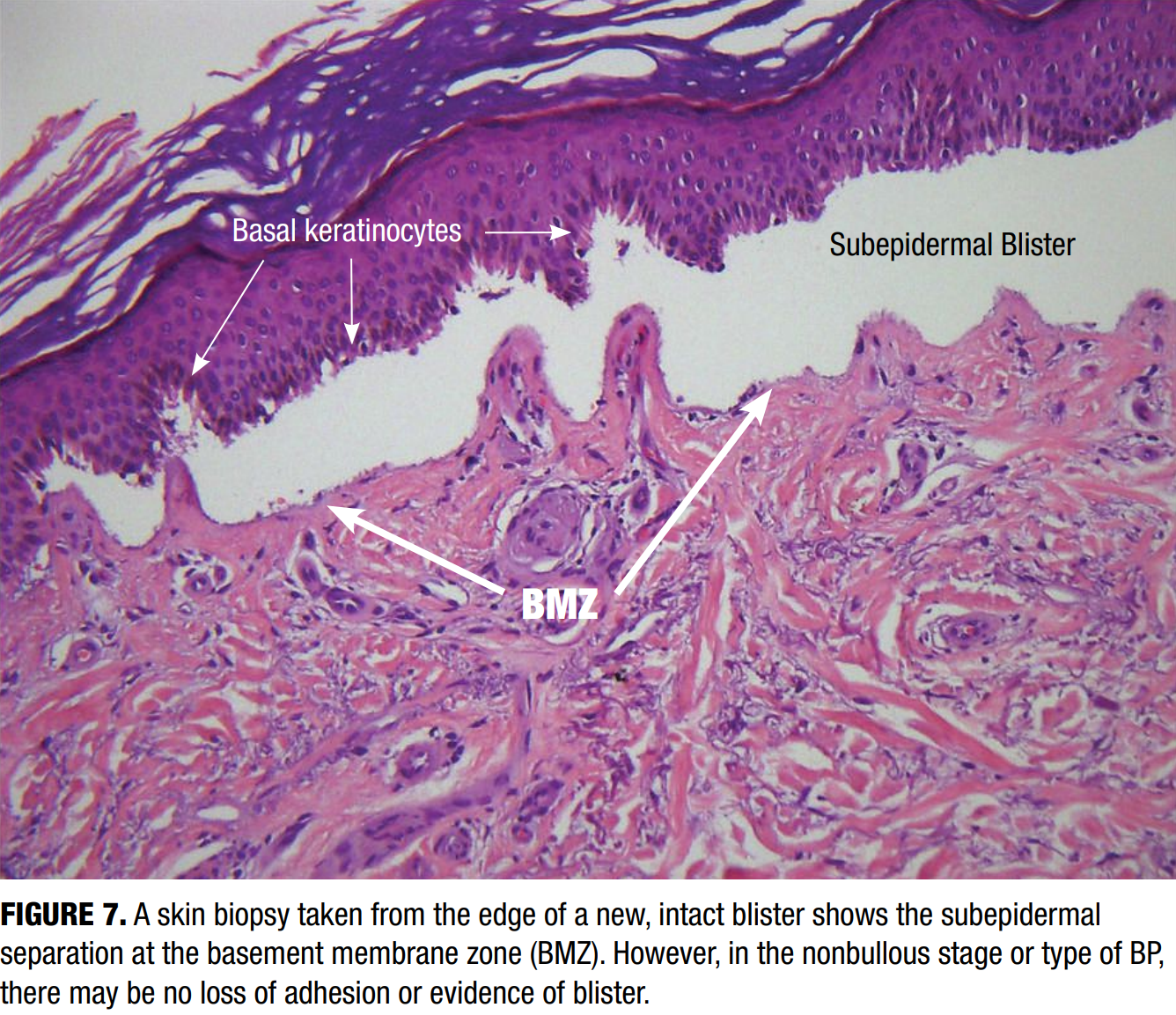
Biopsy for routine histologic examination should be taken from the edge of a fresh blister, ideally submitted in formalin for hematoxylin and eosin (H&E) staining. Characteristic histologic features include a subepidermal separation at the dermoepidermal junction (Figure 7). Subepidermal clefting with a dense eosinophil-rich infiltrate in the superficial dermis can be noted. In early or non-bullous stages, findings may be nonspecific, such as spongiosis, dermal edema, and a mild eosinophilic infiltrate, which can complicate early recognition.26
Direct Immunofluorescence (DIF)
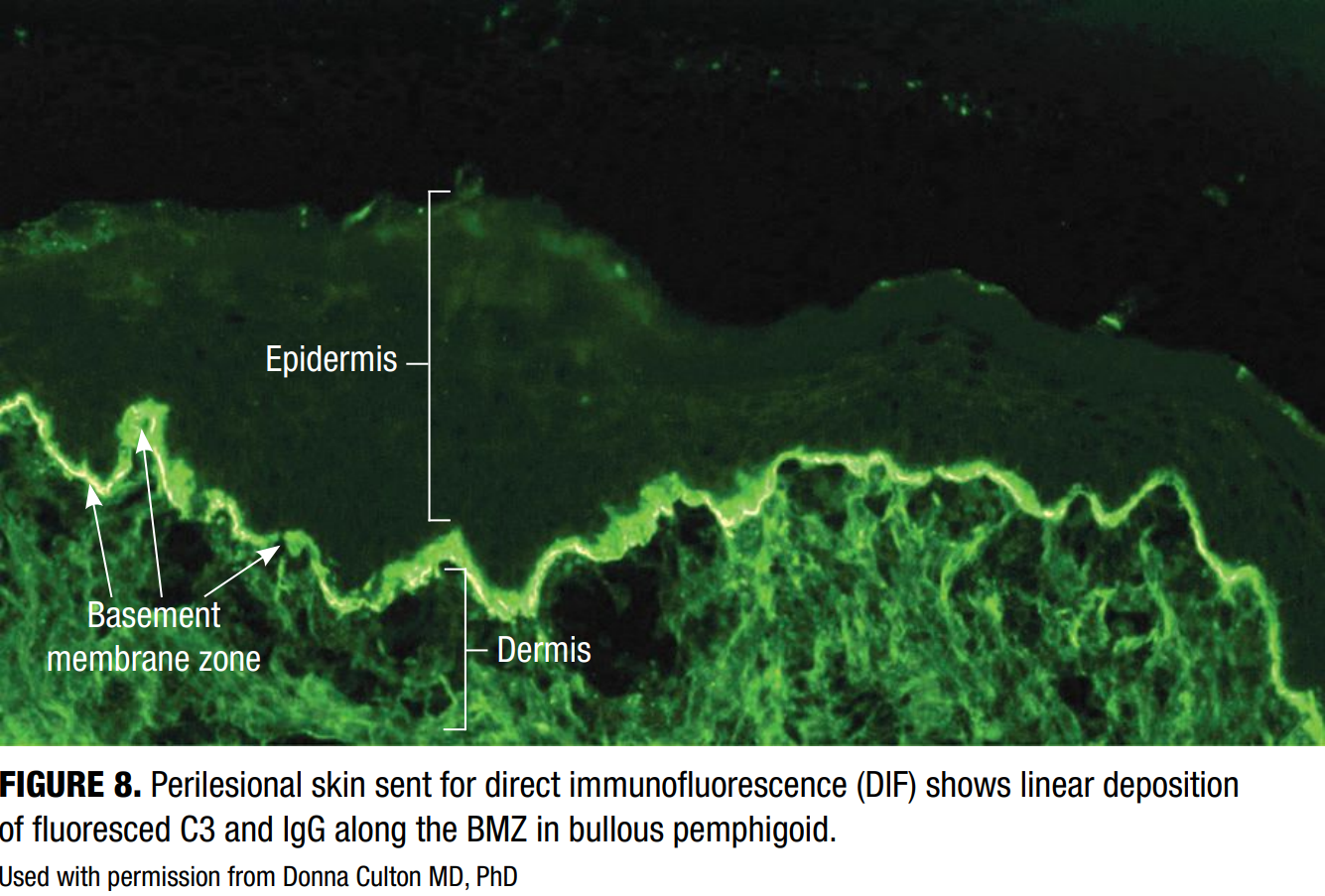
DIF detects in situ deposits of immunoreactants (IgG, IgA, IgM, C3, and fibrin) in perilesional skin using fluorescein-tagged antibodies (Figure 8). In BP, DIF typically reveals linear deposition of IgG and/or C3 along the BMZ.4 The specimen should be obtained from perilesional skin (3–10mm from a new blister) and placed in Michel’s transport medium. Proper specimen collection and transport are essential, as higher risk
of false-negative results may occur if the biopsy is:
- Taken directly from the blister or necrotic tissue
- Located on the lower extremities
- Obtained during the early or urticarial phase of BP
- Inadequately preserved during transport4,5
DIF offers >90% sensitivity and up to 98% specificity when performed appropriately, making it the cornerstone of BP diagnosis.16
Indirect immunofluorescence (IIF)
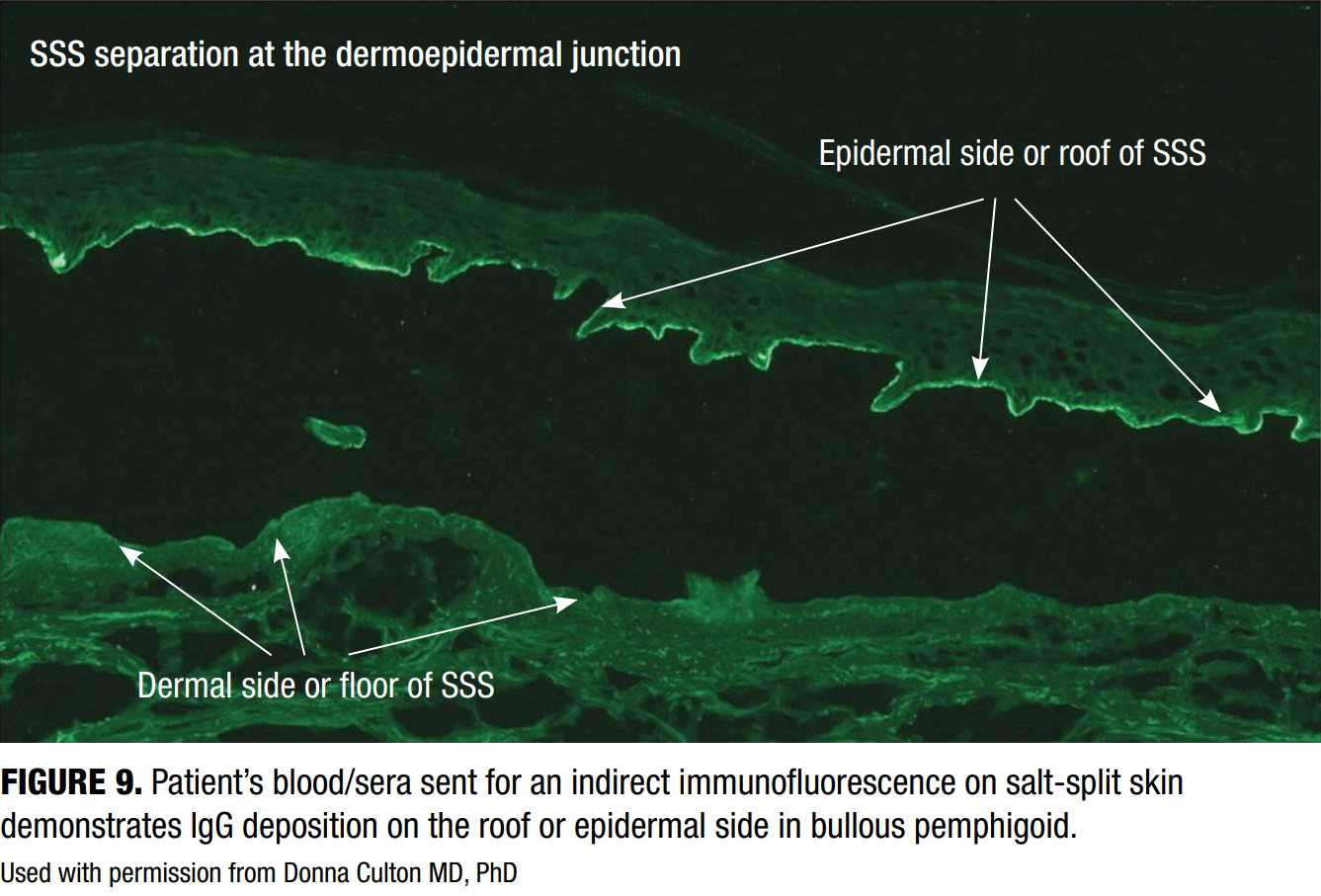
IIF involves incubation of patient serum with a substrate, often salt-split human skin or monkey esophagus, to detect circulating autoantibodies. In BP, IIF shows binding to the epidermal side (or roof) of the salt-split skin (Figure 9), distinguishing it from epidermolysis bullosa acquisita, which binds to the dermal side.20 The binding pattern in the IIF is particularly useful for differentiating BP from other AIBDs.
Enzyme-linked immunosorbent assay (ELISA)
ELISA quantifies serum autoantibodies against BP180 (NC16A domain) and BP230. These tests support diagnosis when used in conjunction with DIF and histology. Anti-BP180 antibodies correlate well with disease activity, making ELISA a useful tool for both diagnosis and monitoring treatment response or detecting impending relapse.12,27 Anti-BP230 antibodies are less specific and are often present in combination with BP180 antibodies.

Accurate diagnosis relies on a combination of clinical evaluation, histopathologic findings, direct immunofluorescence (DIF), indirect immunofluorescence (IIF), and serologic studies.
Conclusion
BP is the most common autoimmune blistering disease in the elderly, with a diverse clinical spectrum that includes non-bullous, drug-associated, mucosal, and gestational variants. Its pathogenesis involves autoantibodies against BP180 and BP230, leading to inflammation and subepidermal blister formation. Comorbidities, especially neurologic and thromboembolic disorders, further complicate disease management and contribute to morbidity.
Accurate diagnosis relies on integrating clinical findings with histopathology, DIF, IIF, and serologic assays. DIF remains the gold standard, while ELISA testing for anti-BP180 aids in both diagnosis and monitoring. A high index of suspicion is essential, particularly in early or atypical presentations.
As BP incidence rises with an aging population, clinicians must remain vigilant, ensure access to appropriate diagnostic resources, and adopt a multidisciplinary approach to improve outcomes and reduce complications.
Currently, BP treatment involves topical corticosteroids for mild cases and systemic corticosteroids or other immunomodulatory agents for moderate to severe cases. The US Food and Drug Administration (FDA) has accepted the supplemental Biologics License Application (sBLA) for dupilumab (Dupixent, Sanofi & Regeneron) for priority review as a potential treatment for adults with BP. The FDA’s decision is expected by June 20, 2025.
If approved, dupilumab would become the first targeted therapy available for BP in the United States.
Stay tuned for Part Two of this series, which will focus on therapeutic management and monitoring of BP.
REFERENCES
- Kridin K, Shihade W, Bergman R. Mortality in patients with bullous pemphigoid: a retrospective cohort study, systematic review and meta-analysis. Acta Derm Venereol. 2019;99(1):72–77. https://pubmed.ncbi.nlm.nih.gov/29963683/
- Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJP, West J. Bullous pemphigoid and pemphigus vulgaris incidence and mortality: population-based cohort study. BMJ. 2008;337(7662):a180. https://pubmed.ncbi.nlm.nih.gov/18614511/
- Marazza G, Pham HC, Schärer L, et al. Incidence of bullous pemphigoid and pemphigus in Switzerland: a 2-year prospective study. Br J Dermatol. 2009;161(4):861–868. https://pubmed.ncbi.nlm.nih.gov/19566661/
- Parker Sareta RS, MacKelfresh J. Autoimmune blistering diseases in the elderly. Clin Dermatol. 2011;29(1):69–79. https://pubmed.ncbi.nlm.nih.gov/21146735/
- Benzaquen M et al. Drug-induced bullous pemphigoid: an update including the emerging role of DPP-4 inhibitors. Am J Clin Dermatol. 2018;19(5):645–653.
- Vassileva S. Drug-induced pemphigoid: bullous and non-bullous variants. Clin Dermatol. 1998;16(3):379–387. https://pubmed.ncbi.nlm.nih.gov/9642531/
- Genovese, G et al. New Insights Into the Pathogenesis of Bullous Pemphigoid: 2019 Update. Frontiers in Immunology. 2019.10:1506. https://pubmed.ncbi.nlm.nih.gov/31312206/
- Di Zenzo G, Della Torre R, Zambruno G, Borradori L. Bullous pemphigoid: from the clinic to the bench. Clin Dermatol. 2012;30(1):3–16. https://pubmed.ncbi.nlm.nih.gov/22137222/
- Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Annu Rev Pathol. 2016;11:175–197. https://pubmed.ncbi.nlm.nih.gov/26907530/
- Schmidt E, Zillikens D. Modern diagnosis of autoimmune blistering skin diseases. Autoimmun Rev. 2010;10(2):84–89. https://pubmed.ncbi.nlm.nih.gov/20713186/
- Saniklidou AH, Tighe PJ, Fairclough LC, Todd I. IgE autoantibodies and their association with the disease activity and phenotype in bullous pemphigoid: a systematic review. Arch Dermatol Res. 2018;310(1):11-28. https://pubmed.ncbi.nlm.nih.gov/29071428/
- Liu Z, Diaz LA, Troy JL, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest. 1993;92(5):2480–2488. https://pubmed.ncbi.nlm.nih.gov/7693763/
- Edwards G, Diercks GFH, Seelen MAJ,et al. Complement activation in autoimmune bullous dermatoses: a comprehensive review. Front Immunol. 2019.26;10:1477 https://pmc.ncbi.nlm.nih.gov/articles/PMC6606728/
- Chen F, Wang Y, Chen X, Yang N, Li L. Targeting interleukin 4 and interleukin 13: a novel therapeutic approach in bullous pemphigoid. Ann Med. 2023;55(1):1156–1170 https://pubmed.ncbi.nlm.nih.gov/36999962/
- Arakawa M, Dainichi T, Ishii N,et al. Lesional Th17 cells and regulatory T cells in bullous pemphigoid. Exp Dermatol. 201;20(12):1022–1024. https://pubmed.ncbi.nlm.nih.gov/22017210/
- Clapé A, Muller C, Gatouillat G,et al. Mucosal involvement in bullous pemphigoid Is mostly associated with disease severity and to absence of anti-BP230 autoantibody. Front Immunol. 2018;9:479. https://pubmed.ncbi.nlm.nih.gov/29662486/
- Cordel N, Chosidow O, Hellot MF, et al. Neurological disorders in patients with bullous pemphigoid. Dermatology. 2007;215(3):187–191. https://pubmed.ncbi.nlm.nih.gov/17823513/
- ilani-Nejad N, Zhang M, Kaffenberger J. The association between bullous pemphigoid and neurological disorders: a systematic review. Eur J Dermatol. 2017;27(5):472–481. https://pubmed.ncbi.nlm.nih.gov/28681724/
- Cugno M, Marzano AV, Bucciarelli P, et al. Increased risk of venous thromboembolism in patients with bullous pemphigoid. The INVENTEP (INcidence of VENous ThromboEmbolism in bullous Pemphigoid) study. Thromb Haemost. 2016 Jan;115(1):193-9. https://pubmed.ncbi.nlm.nih.gov/26245987/
- Meijer JM, Diercks GFH, de Lang EWG, et al. Assessment of diagnostic strategy for early recognition of bullous and nonbullous variants of pemphigoid. JAMA Dermatol. 2019;155(2):158–165. https://pubmed.ncbi.nlm.nih.gov/30624575/
- Verheyden MJ, Bilgic A, Murrell DF. A systematic review of drug-induced pemphigoid. Acta Derm Venereol. 2020;100(15): adv00224. https://pubmed.ncbi.nlm.nih.gov/32176310/
- Du G, Patzelt S, van Beek N, Schmidt E. Mucous membrane pemphigoid. Autoimmun Rev. 2022;103036.https://pubmed.ncbi.nlm.nih.gov/34995762/
- Shimanovich I, Bröcker EB, Zillikens D. Pemphigoid gestationis: new insights into the pathogenesis lead to novel diagnostic tools. BJOG. 2002;109(9):970–976.https://pubmed.ncbi.nlm.nih.gov/12269691/
- Abdelhafez M, Ahmed KAM, Daud MNBM, et al. Pemphigoid gestationis and adverse pregnancy outcomes: A literature review. J Gynecol Obstet Hum Reprod. 2022;51(5):102370. https://pubmed.ncbi.nlm.nih.gov/35385801/
- Sävervall C, Sand FL, Thomsen SF. Dermatological diseases associated with pregnancy: pemphigoid gestationis, polymorphic eruption of pregnancy, intrahepatic cholestasis of pregnancy, and atopic eruption of pregnancy. Dermatol Res Pract. 2015:2015:979635.https://pubmed.ncbi.nlm.nih.gov/26609305/
- Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. 2017;18(4):513–528. https://pubmed.ncbi.nlm.nih.gov/28247089/
- Zillikens D, Mascaro JM, Rose PA, et al. A highly sensitive enzyme-linked immunosorbent assay for the detection of circulating anti-BP180 autoantibodies in patients with bullous pemphigoid. J Invest Dermatol. 1997;109(5):679-83. https://pubmed.ncbi.nlm.nih.gov/9347799/
ABOUT THE AUTHOR
Margaret Bobonich, DNP, FNP-C, DCNP, FAANP, is a Nurse Practitioner and the Founder of Center for Dermatology NPs and PAs. She is also the founding Co-Editor-in-Chief of JDNPPA.
DISCLOSURES
Margaret Bobonich is on the Advisory Boards of Lilly USA, Bristol Myers Squibb, Organon (Dermavant), and Janssen. She is a Consultant for Lilly USA, Organon (Dermavant), and Bristol Myers Squibb. Bobonich also serves on the Speaker’s Bureaus for Sanofi and Regeneron.
