
By Cheryl Guttman Krader | Reviewed by and from the files of Joseph Kamel, DO, and Neil Korman, MD, PhD
CASE HISTORY
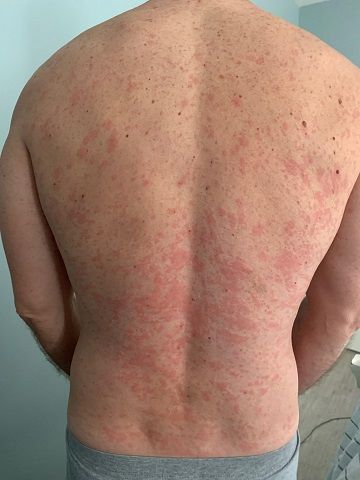
A 46-year-old Caucasian man presented with a 3-year history of a widespread pruritic evanescent eruption (Figures 1 A-C). He had previously seen dermatologists and allergists who had diagnosed idiopathic chronic spontaneous urticaria.
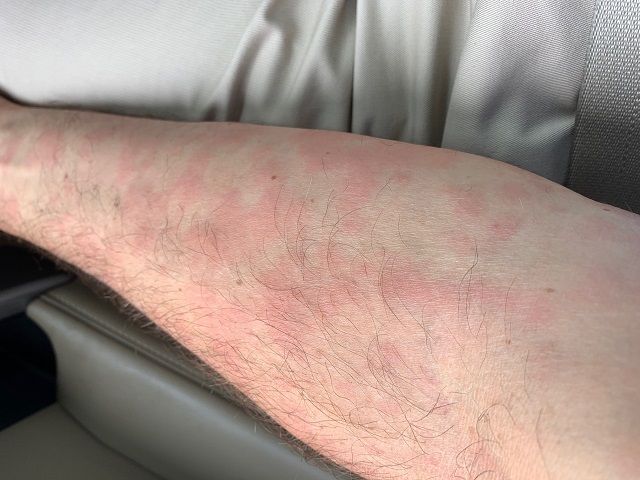
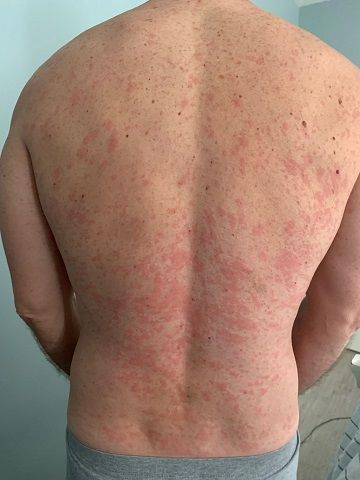
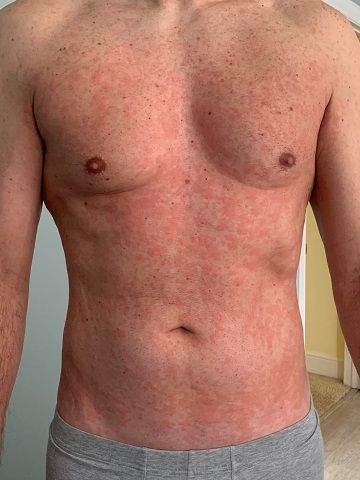
The patient denied any episodes of angioedema and any medication use including OTC supplements. He denied any correlation with temperature changes, exercise, pressure, exposure to sunlight, or water, though he did report a burning sensation during warm-water showers. The personal and family medical history were both unremarkable. Prior treatments included numerous second-generation antihistamines with good response but in the recent several months the eruption had become refractory to therapy. For the past 6 months, the patient had developed bilateral leg weakness, intermittent night sweats, cyclic low-grade fevers (up to 100.8° F), arthralgias, and myalgias. He had responded to pulsed oral prednisone, but in the past 2 months had lost all response to prednisone.
A skin biopsy performed when the systemic symptoms developed showed perivascular and interstitial neutrophils, perivascular lymphocytes, and scattered eosinophils. Direct immunofluorescence was negative for vasculitis.
Laboratory studies showed lymphopenia and monocytosis, with elevated M-protein, γ globulin, α-1 globulin, α-2 globulin, IgM, C-reactive protein, and erythrocyte sedimentation rate. Liver and renal function tests were normal. Flow cytometry was negative for clonal lymphoid proliferation, and FISH was negative for BCR-ABL translocation.
A bone marrow biopsy showed 1% plasma cells and mildly hypercellular bone marrow with trilineage hematopoiesis and myeloid predominance. Abdominal ultrasound showed only reactive inguinal lymphadenopathy; lymphadenopathy was absent on physical examination. Skeletal CT scan showed mild degenerative changes with no focal bony abnormalities or lytic lesions.
What is your suspected diagnosis?
The patient was diagnosed with Schnitzler syndrome using Strasbourg criteria (Table).1 He was started on off-label treatment with the interleukin-1 (IL-1) receptor antagonist anakinra (Kineret) 100 mg subcutaneous daily. Follow-up is pending.
DISCUSSION
The differential diagnosis for chronic spontaneous urticaria is broad. Since most cases are idiopathic, in the absence of history or clinical findings that raise suspicion for another diagnosis a limited initial workup is recommended, and treatment with a second-generation antihistamine is recommended.2 The refractory nature of the urticaria in this patient combined with the development of systemic symptoms spurred a more intensive diagnostic evaluation.
Entities to consider in the differential diagnosis for treatment-refractory chronic urticaria accompanied by fever, arthralgia, and increased inflammatory parameters include cryopyrin-associated periodic disease (CAPS), adult-onset Still’s disease (AOSD), urticarial vasculitis, Sweet syndrome, and Schnitzler syndrome. CAPS develops in childhood, the rash involves the face and is exacerbated by cold, and patients may have hearing loss. AOSD also generally occurs in younger adults and is differentiated by elevated ferritin levels, transaminitis, and pharyngitis. The patient had no morphological or histological evidence of vasculitis.
Schnitzler syndrome
Schnitzler syndrome is a rare, disabling condition that was first described in 1972 by French dermatologist Liliane Schnitzler.3 Consistent with this patient’s history, typical onset is in the fifth decade of life.4
Chronic urticaria may be the first clinical sign.4,5 The eruption typically involves the trunk and limbs and precedes systemic symptoms by several years.4,5 In addition to unexplained recurrent fevers, arthralgias and myalgia, other common clinical signs include asthenia, unintentional weight loss, axillary or inguinal lymphadenopathy, hepatosplenomegaly, and headache.5,6
Schnitzler syndrome generally follows a chronic, non-malignant course. However, 10% to 15% of patients progress to develop a lymphoproliferative disorder, primarily Wäldenstrom macroglobulinemia; median time to development of a malignancy was 8 years in a series of 35 patients.4
Etiology and pathophysiology
Schnitzler syndrome is believed to be an autoimmune disorder with an innate immune-mediated mechanism.5,6 Interleukin-1β (IL-1β) is thought to have a pathogenic role based on the effectiveness of anakinra for inducing remission and considering the finding in some patients of a mosaicism in NLRP3 in myeloid cell lines, which is a gene associated with increased IL-1 activity in monocytes.7,8
Diagnostic work-up
Diagnosis of Schnitzler syndrome is often delayed because chronic urticaria may be the first and only clinical sign for years.4 Protein electrophoresis to identify a monoclonal gammopathy differentiates Schnitzler syndrome from related inflammatory diseases and should be performed in patients with chronic urticarial rash accompanied by clinical and laboratory signs of systemic inflammation.5,6 IgM gammopathy is seen more often than IgG gammopathy in patients with Schnitzler syndrome.6
Management
As seen in this case, antihistamines are generally ineffective for treating the urticarial rash associated with Schnitzler syndrome, and steroids are only moderately effective.6 Anti-IL-1 therapy with anakinra is recommended as first-line treatment for Schnitzler syndrome.1 Because it is highly and rapidly effective for inducing complete remission, some believe that the diagnosis of Schnitzler syndrome should be reconsidered in patients who do not respond to anakinra.5 Anakinra is not curative, however, and symptoms recur soon after treatment stops.4
Efficacy in treating Schnitzler syndrome has also been reported with off-label use of the human IL-1β monoclonal antibody canakinumab (Ilaris).4 Canakinumab is injected subcutaneously every 4 weeks and is associated with fewer injection-site reactions than is anakinra.9
The effect of anti-IL-1 therapy on the risk for developing a lymphoproliferative disorder is unknown. Bone marrow biopsy in this patient ruled out current hematologic malignancy, but ongoing follow-up with oncology is necessary because a minority of patients may develop a hematologic malignancy.
References
1. Simon A, Asli B, Braun-Falco M, et al. Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy. 2013;68(5):562-568.
2. Zuberbier T, Bernstein JA. A comparison of the United States and international perspective on chronic urticaria guidelines. J Allergy Clin Immunol Pract. 2018;6(4):1144-1151.
3. Schnitzler L. Lésions urticariennes chroniques permanentes (érythème pétaloïde?) Cas cliniques no 46B. Journée Dermatologique d’Angers. 1972.
4. de Koning HD. Schnitzler’s syndrome: lessons from 281 cases. Clin Transl Allergy. 2014;4:41.
5. Gusdorf L, Asli B, Barbarot S, et al. Schnitzler syndrome: validation and applicability of diagnostic criteria in real-life patients. Allergy. 2017;72(2):177-182.
6. Davis MDP, van der Hilst JCH. Mimickers of urticaria: urticarial vasculitis and autoinflammatory diseases. J Allergy Clin Immunol Pract. 2018;6(4):1162-1170.
7. Nham T, Saleh C, Chu D, et al. Refractory urticaria and the importance of diagnosing Schnitzler’s syndrome. BMJ Case Rep. 2019;12(4):e228546.
8. de Koning HD, van Gijn ME, Stoffels M, et al. Myeloid lineage-restricted somatic mosaicism of NLRP3 mutations in patients with variant Schnitzler syndrome. J Allergy Clin Immunol. 2015;135(2):561-564.
9. Rossi-Semerano L, Fautrel B, Wendling D, et al; MAIL1 (Maladies Auto-inflammatoires et Anti-IL-1) study group on behalf of CRI (Club Rhumatisme et Inflammation). Tolerance and efficacy of off-label anti-interleukin-1 treatments in France: a nationwide survey. Orphanet J Rare Dis. 2015 Feb 15;10:19.
